因为目前合并多个测序、芯片数据集这一块并没有完全统一的标准,方法大概有五六种。公说公有理婆说婆有理,对于我这样的新手来说,最简单的是跟随顶级文章的文章思路或者分析流程和步骤。
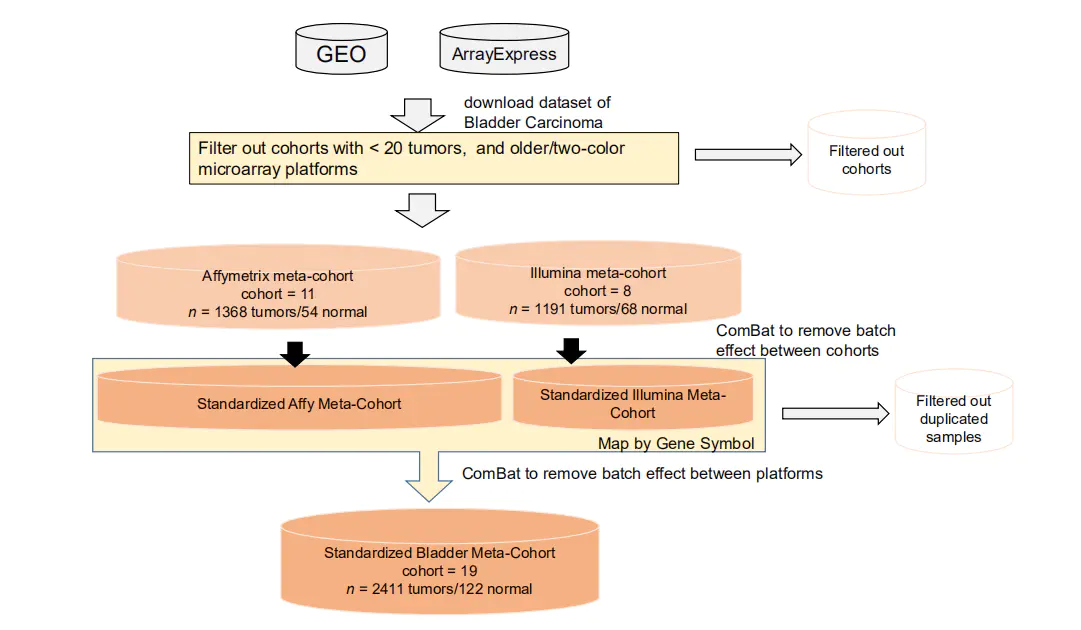
于是我选取了一篇欧洲泌尿外科的顶级文章,从这篇文章的补充材料可以看出来:
移除批次效应前
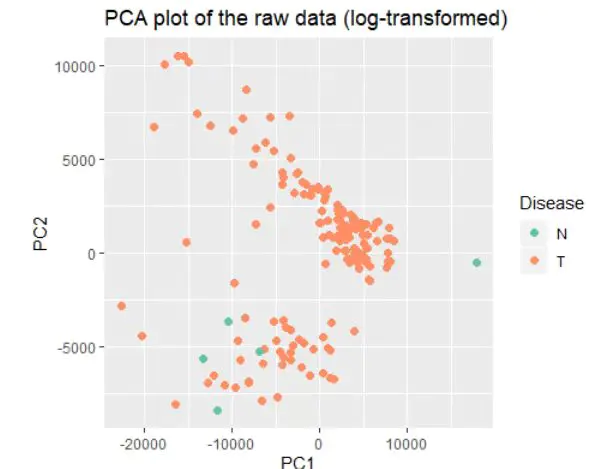
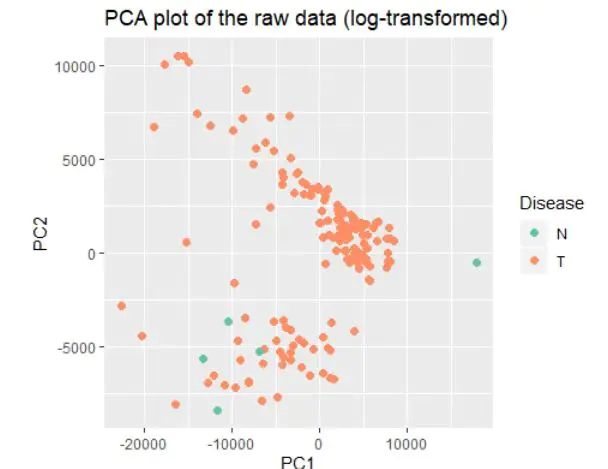
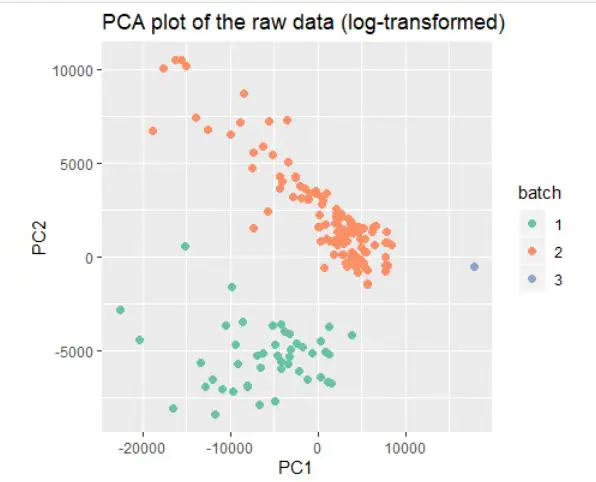

移除批次效应后

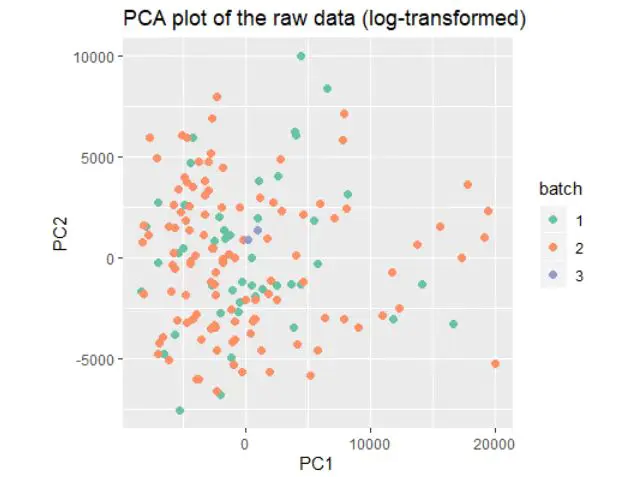
作者将所有的芯片测序数据分为affy和illumina两类;
将affy公司的芯片通过标准的affy公司流程分析及combat;
illumina同样是通过标准的illumina公司流程分析及combat;
最后将两类芯片combat到一起,上一步是消除batch effect是不同的cohort,而这一步是affy和illumina两个平台的区别。
加载包
###step1 加载包################################
# =======================================================
# if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#
# BiocManager::install("bladderbatch")
rm(list=ls())
library("sva")
options(stringsAsFactors=FALSE)
setwd('D:\\train\\data')
library(bladderbatch)
加载用于分析的数据
# =======================================================
###step2 加载用于分析的数据################################
#包括一个表达量矩阵和一个分组文件
# =======================================================
data(bladderdata)
edata = exprs(bladderEset)
edata[1:5,1:5]
pheno = pData(bladderEset)
pheno[1:5,1:4]
dt <- data.frame(cel=rownames(pheno), pheno)
dt[1:5,1:5]
绘制图片显示以前聚类效果
# =======================================================
###step3 绘制图片显示合并以前的聚类结果##################
# =======================================================
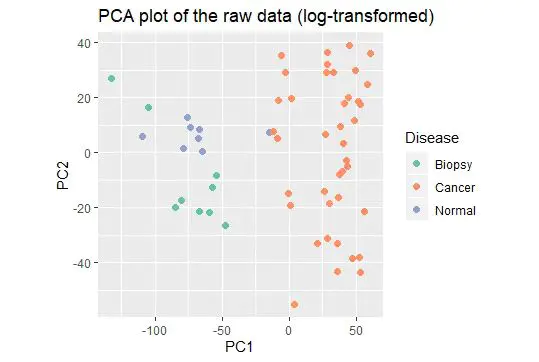
PCA_raw <- prcomp(t(edata), scale = FALSE)
dataGG <- data.frame(PC1 = PCA_raw$x[,1], PC2 = PCA_raw$x[,2],
Disease = dt$cancer,
batch = dt$batch)
dataGG$batch <- as.factor(dataGG$batch)
library(ggplot2)
(qplot(PC1, PC2, data = dataGG, color = Disease,
main = "PCA plot of the raw data (log-transformed)", size = I(2),
asp = 1.0)
+ scale_colour_brewer(palette = "Set2"))
library(ggplot2)
(qplot(PC1, PC2, data = dataGG, color = batch,
main = "PCA plot of the raw data (log-transformed)", size = I(2),
asp = 1.0)
+ scale_colour_brewer(palette = "Set2"))
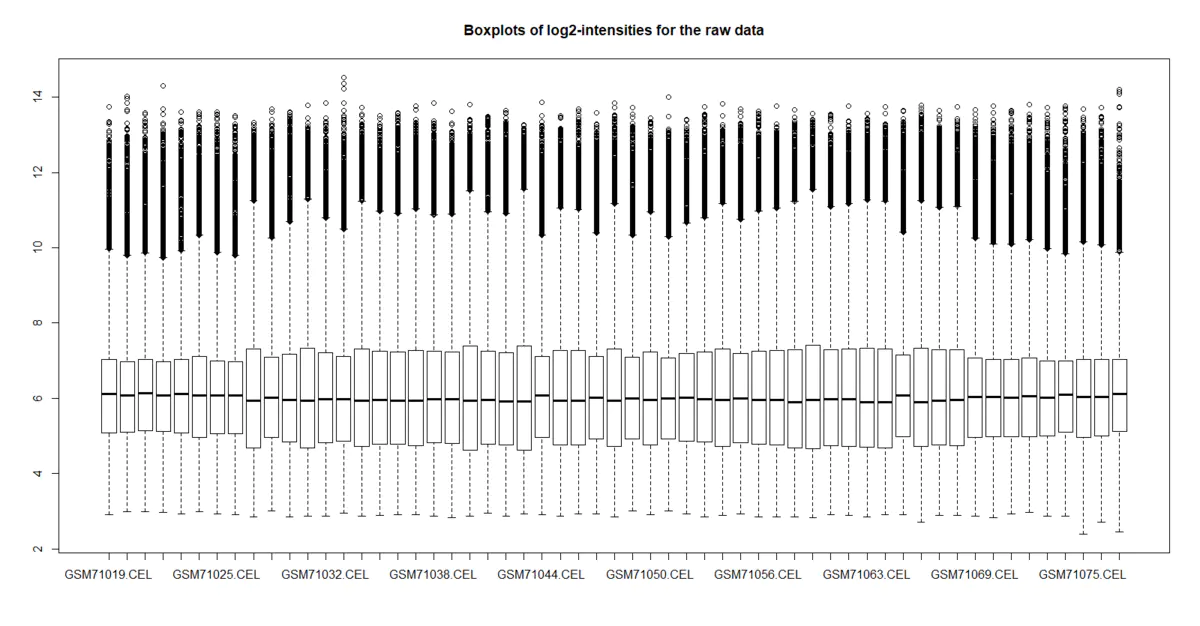
boxplot(edata, target = "core",
main = "Boxplots of log2-intensities for the raw data")
dist_mat <- dist(t(edata))
clustering <- hclust(dist_mat, method = "complete")
par(mfrow=c(2,1))
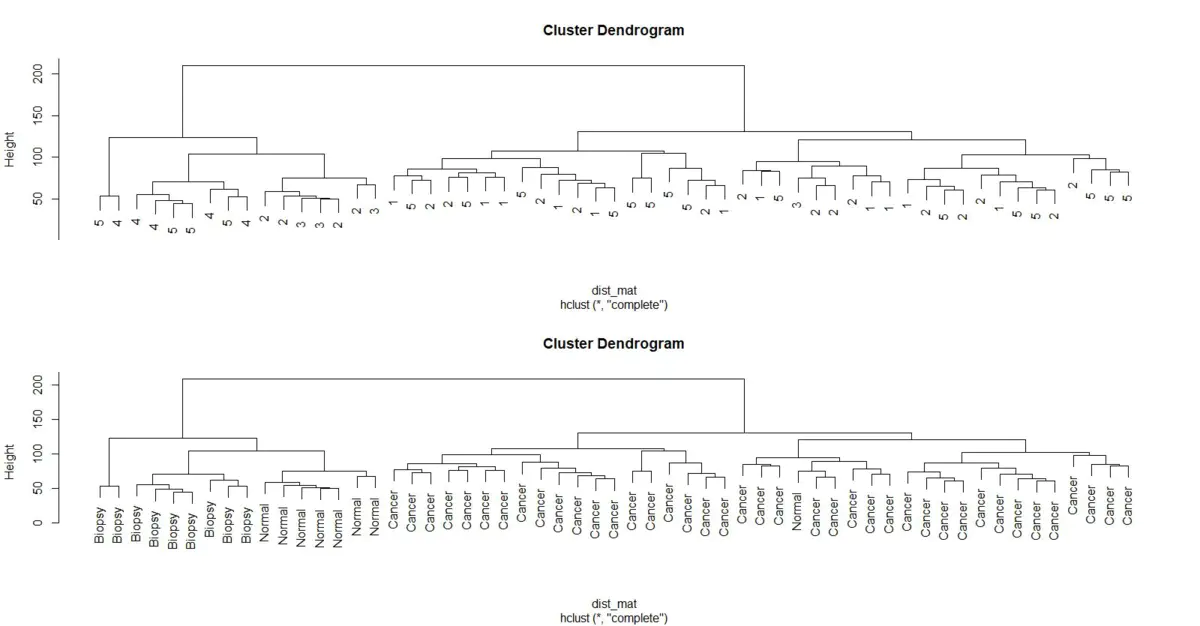
plot(clustering, labels = pheno$batch)
plot(clustering, labels = pheno$cancer)

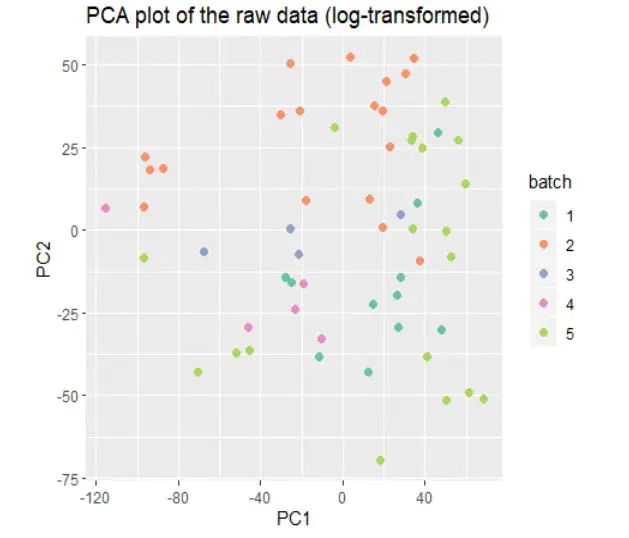
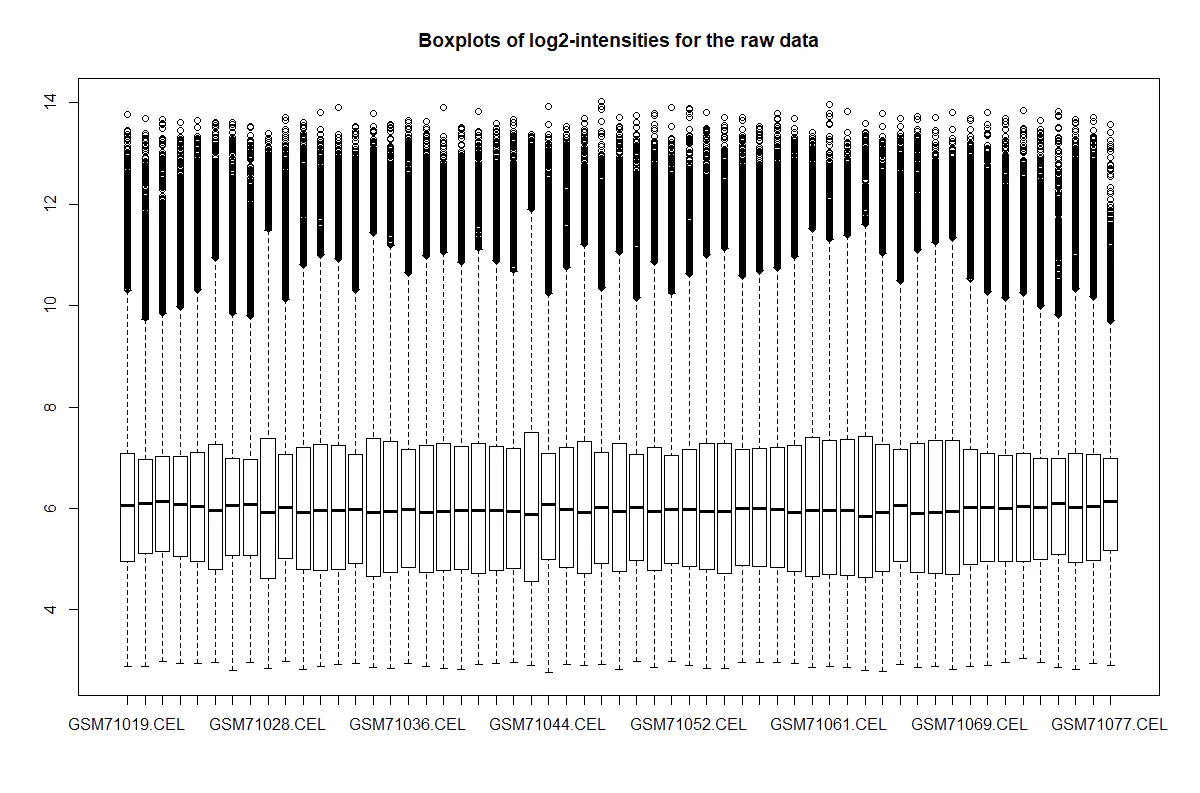
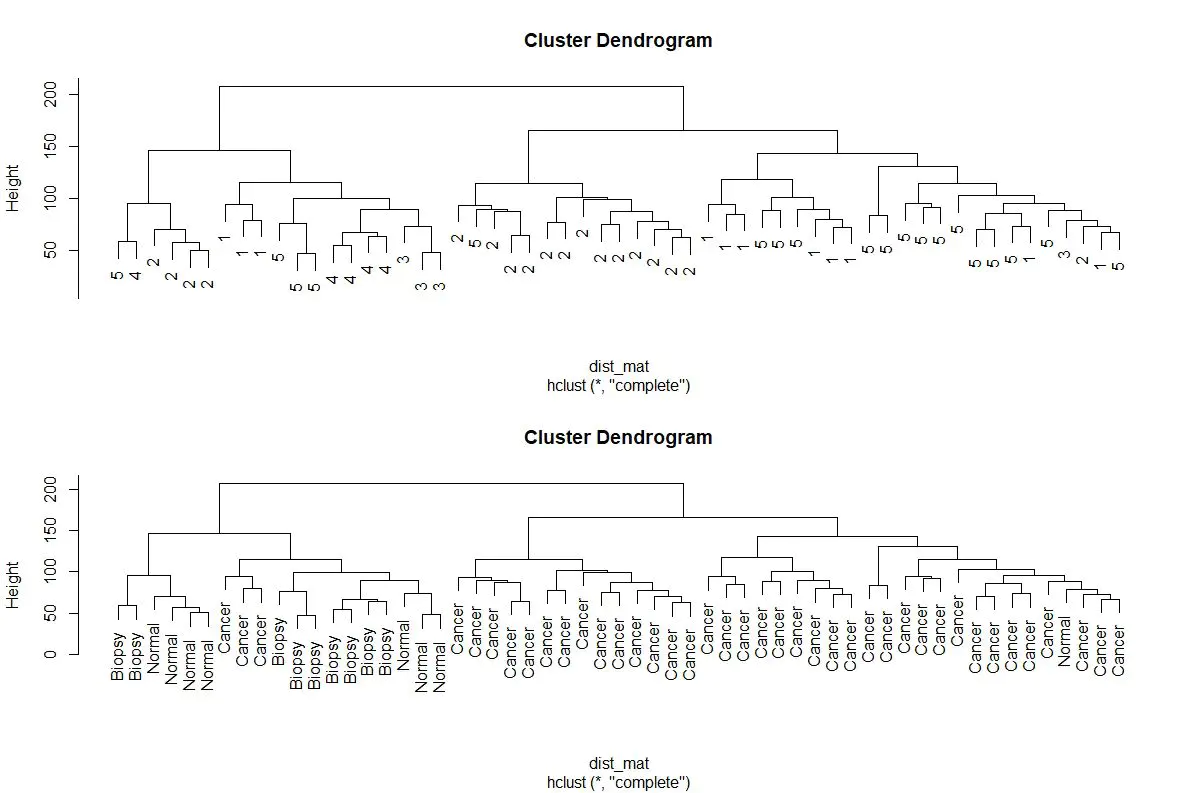
从这些图片看出有明显批次效应,批次效应出现在以下情形:
一个实验不同部分在不同时间完成;
一个实验不同部分不同人完成;
试剂用量、芯片、实验仪器不同;
自己的数据与网站数据混用
Combat参数(prarametric)或者非参数(non-prarametric)的经验贝叶斯框架进行批次效应校正。
移除批次效应
1 | # ======================================================= |
绘制聚类后的图片
1 |
|
前后对比,可以发现不同batch基本不再是泾渭分明,而我们呢关心的cancer和normal泾渭分明了,便于下一步分析。

实例验证 数据
1 |
|
question:
同属于illumina平台的batch1和batch2的移除批次效应结果很满意,但是affy效果一般;
1.可能是affy数据表达矩阵为原始数据,而illumina为标准化后的数据,所以差距大;
2.这篇顶刊提示我么为什么对affy标准化后才和illumina合并